


INTRODUCTION
Les leucémies aiguës myéloblastiques (ou myéloïdes) sont des hémopathies aiguës caractérisées par un blocage de maturation des lignées médullaires myéloïdes normales avec envahissement progressif de la moelle, du sang, des organes hématopoïétiques, et éventuellement des organes non hématopoïétiques par des éléments jeunes anormaux appelés myéloblastes.
Ces éléments vont inhiber la croissance des lignées myéloïdes normales d’où la coexistence d’un syndrome tumoral dû à la prolifération myéloblastique et d’un syndrome d’insuffisance médullaire.
I - CIRCONSTANCES ETIOLOGIQUES DU DIAGNOSTIC
A - AGE ET SEXE :
La fréquence des leucémies myéloblastiques est d’autant plus importante que l’âge est plus avancé. La médiane d’âge se situe entre 60 et 65 ans. L’incidence annuelle est d’environ 4 pour 100 000 après 60 ans. Les deux sexes sont atteints de manière sensiblement égale.
B - CIRCONSTANCES FAVORISANTES :
Elles sont retrouvées dans environ 15 % des cas. Leurs présences aggravent le pronostic.
1 - Circonstances professionnelles :
La leucémie doit faire l’objet d’une déclaration en tant que maladie professionnelle lorsque les expositions suivantes sont retrouvées :
Radiations ionisantes : rayons X ou gamma
Benzène et ses dérivés
2 - Agents chimiothérapiques :
On observe une fréquence de 3 à 6 % des leucémies aiguës secondaires après un cancer curable par chimiothérapie. Les agents principalement en causes sont les alkylants et les nitroso-urées. Le délai médian entre le début de la chimiothérapie et l’apparition de la leucémie aiguë myélobastique secondaire est de 5 ans.
II - DIAGNOSTIC POSITIF
A - SIGNES CLINIQUES :
La leucémie aiguë myéloblastique peut être découverte lors d’un hémogramme systématique, mais elle est le plus souvent découverte devant l’apparition sur quelques jours à quelques semaines de signes cliniques d’insuffisance médullaire et/ou un syndrome tumoral réalisant parfois un tableau dramatique d’emblée.
1 - Syndrome d’insuffisance médullaire :
Signes fonctionnels d’anémie: Essoufflement, palpitation, lipothymie, céphalées, pâleur, dyspnée d’effort.
Infections: Elles sont liées à la neutropénie et attirent souvent l’attention. Elles sont fréquentes en particulier au niveau de la sphère ORL. La fièvre peut aussi être spécifiquement leucémique.
Hémorragies: Elles sont liées à la thrombopénie et éventuellement à une coagulopathie. Elles se manifestent en général uniquement par des pétéchies cutanées, des gingivorragies lors du brossage des dents et des épistaxis. Des hémorragies plus graves peuvent cependant être révélatrices: hémorragies digestives, hémorragies cérébro-méningées.
2 - Syndrome tumoral :
Il est surtout retrouvé dans les formes avec un composant monoblastique :
Hépatosplénomégalie
Adénopathies superficielles
Gingivite hypertrophique
Localisations cutanées : elles réalisent en général des papules rougeâtres appelées leucémides.
Envahissement méningé: rare.
Douleurs osseuses intenses.
3 - Syndrome de leucostase :
Ce syndrome est lié à l’hyperviscosité sanguine observée en cas d’hyperleucocytose considérable (supérieure à 100 G/l). Sur le plan clinique, ce syndrome peut s’extérioriser par une dyspnée alors que la radio pulmonaire montre peu d’anomalies (opacités diffuses bilatérales), des céphalées et une obnubilation traduisant la mauvaise circulation au niveau des capillaires pulmonaires et cérébraux.
B – BIOLOGIE :
1 - Syndrome Hématologique :
L’étude cytologique permet de confirmer le diagnostic.
La leucoblastose est variable et peut être inexistante au niveau du sang périphérique. Elle est parfois considérable et entraîne un risque vital immédiat si elle est supérieure à 100 G/l. La leucocytose globale est surtout fonction de la leucoblastose.
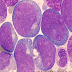
Myélogramme : Il est l’examen fondamental pour le diagnostic. Il permet de classer cytologiquement la leucémie ainsi que des études complémentaires cytochimiques, cytogénétiques, immunologiques et moléculaires. 1-Classification FAB
¨ M0 : indifférenciée avec des blastes de grande taille sans granulation, MPO - et Noir-Soudan -. L’étude immunologique est en faveur de l’origine myéloïde.
 |
| LAM0 |
¨ M1 : myéloblastique indifférenciée (15% des cas). Les blastes représentent au moins 90% des cellules non érythroïdes. Il n’y a pas de maturation et peu de granulations.
 |
| LAM1 |
¨ M2 : myéloblastique avec maturation myéloïde partielle des éléments (20% des cas). Les blastes représentent 20 à 89% des cellules non érythroïdes avec persistance d’une maturation jusqu’au promyélocyte. Les cellules sont généralement nucléolées avec des granulations azurophiles et des bâtonnets d’Auer.
 |
| LAM2 |
¨ M3 : promyélocytaire (8% des cas) où les granulations péroxydases positives sont extrêmement nombreuses pouvant couvrir le noyau des leucoblastes. Les corps d’Auer peuvent parfois se grouper en amas ou fagots. Il existe un sous-type variant (M3v) correspondant à une forme hypogranulocytaire. L’abondance des granulations riches en activateurs de la coagulation est responsable de la coagulopathie observée de manière pratiquement constante dans cette forme. La forme promyélocytaire touche surtout les sujets jeunes. Il existe souvent une pancytopénie périphérique.
 |
| LAM3 |
¨ M4 : myélo-monoblastique (25% des cas) avec coexistence de myéloblastes et de monoblastes reconnus par leur noyau volontiers plus contourné contenant de nombreux nucléoles et la présence à leur niveau d’estérases inhibées par le chlorure de sodium. Il existe parfois des éosinophiles anormaux (> 5%) ou des basophiles anormaux.
 |
| LAM4 |
¨ M5 : monoblastique (25% des cas), avec présence uniquement de monoblastes. Il existe 2 sous-types: une forme peu différenciée (M5a) et une forme différenciée (M5b). La leucoblastose périphérique peut être importante. Il existe souvent des localisations extra-médullaires en particulier gingivales et cutanées.
 |
| LAM5 |
¨ M6 : érythroblastique (8% des cas) avec érythroblastose supérieure à 50% des éléments médullaires et un contingent blastique souvent plus modéré.
 |
| LAM6 |
¨ M7 : mégacaryoblastique (1% des cas) reconnue par la présence d’excroissances cellulaires en périphérie du cytoplasme des cellules leucémiques évoquant une plaquetopoïèse anormale. Surtout la microscopie électronique montre les péroxydases plaquettaires et permet d’en faire un diagnostic de certitude.
 |
| LAM7 |
2-Classification OMS 2008
LAM avec anomalies génétiques récurrentes
LAM liées à des anomalies myélodysplasiques
LAM thérapies induites
LAM NOS
Sarcome myéloïde
Proliférations myéloïdes liées aux syndrome de Down
Leucémies à cellules dendritiques blastiques plasmocytoides
a-LAM avec anomalies génétiques récurrentes:
LAM avec t(8;21)
LAM avec inv/del16
LAM avec t(15;17)
LAM avec t(9;11)
LAM avec t(6;9)
LAM avec inv(3)
LA mégacaryoblastique avec t(1;22)
LAM avec mutation
b-LAM avec anomalie associée aux SMD:
Critères Dg:
20 % de blastes sanguins ou médullaires
Et ATCD de SMD ou anomalie génétique
liée à un SMD ou dysplasie multignée
Et absence de radiothérapie ou
chimiothérapie antérieure
Pas d’anomalies cytogénétiques
récurentes.
III - DIAGNOSTIC DIFFERENTIEL
A - AFFECTION NON MALIGNE :
1 - Agranulocytose en voie de récupération :
La moelle est riche en éléments myéloïdes jeunes, mais restent assez polymorphe sans atteinte des autres lignées, l’évolution se fait rapidement vers la normalisation
2 - Déficit vitaminique :
Ils peuvent entraîner une pancytopénie à moelle riche avec troubles de maturation mais il existe une mégaloblastose.
B - AUTRES HEMOPATHIES MALIGNES :
1 - Leucémie aiguë lymphoblastique :
Elle pose un problème de diagnostic uniquement dans les formes indifférenciées. L’immunologie a montré qu’environ 10 % des leucémies aiguës étaient en fait des formes frontières à différentiation mixte myéloïde et lymphoïde et associée à un pronostic particulièrement sombre.
2 - Myélodysplasies en cours de transformation :
Il s’agit là surtout de problèmes nosologiques et par définition, dans ces cas, le pourcentage des blastes médullaires est inférieur à 30 %.
IV - FORMES CLINIQUES
Les formes granuleuses (M1, M2, M6) sont souvent peu tumorales. Rarement on rencontre des tumeurs localisées. Dans les formes hyperleucocytaires, il peut exister une leucostase surtout pulmonaire et une coagulopathie.
Les formes promyélocytaires (M3) sont généralement non tumorales et peu hyperleucocytaires. Les complications hémorragiques liées à la coagulopathie sont fréquentes. Dans la forme variante (M3v), le taux de globules blancs est souvent très élevé.
Les formes monoblastiques (M4 et surtout M5) sont souvent hyperleucocytaires et tumorales. On y note des localisations cutanées et gingivales. L’envahissement méningé y est plus fréquent que dans les autres types de LAM.
V - EVOLUTION ET TRAITEMENT
L’évolution de la leucémie aiguë myéloblastique non traitée est mortelle en quelques semaines. Son diagnostic impose l’hospitalisation dans un service spécialisé pour mettre en route un traitement.
A - INDUCTION DE LA REMISSION COMPLETE :
Le principe de la chimiothérapie d’induction est d’abaisser le plus rapidement possible la masse de cellules leucémiques au prix d’une aplasie médullaire de 2 à 4 semaines après la fin du cycle d’induction lui-même de 5 à 10 jours. La rémission complète est définie par une numération formule normale et un myélogramme contenant moins de 5% de myéloblastes.
1 – Chimiothérapie :
Il s’agit d’une polychimiothérapie associant le plus souvent deux types de produits :
Les anthracyclines et leurs dérivés (daunorubicine, idarubicine, mitoxantrone) sont des agents intercalants à toxicité hématologique, digestive, sur le système pileux, et cardiaque à doses cumulatives justifiant la pratique systématique d’un électrocardiogramme et éventuellement d’une échographie ou d’une scintigraphie cardiaque avant traitement. Leur utilisation est contre-indiquée en cas de cardiopathie décompensée.
La cytosine arabinoside est un analogue structural des bases puriques à toxicité hématologique, digestive, et sur le système pileux. A très fortes doses, on peut observer également une toxicité cérébelleuse.
B - MAINTIEN DE LA REMISSION COMPLETE :
A l’issue de l’aplasie consécutive à l’induction de la rémission, un myélogramme va être réalisé et si celui-ci affirme la rémission complète, une thérapeutique sera envisagée pour éviter les rechutes.
1 - Chimiothérapie avec ou sans autogreffe de cellules souches hématopoïétiques :
On peut employer soit des cures de chimiothérapie intensives et aplasiantes nécessitant de nouvelles hospitalisations (chimiothérapies de consolidation), soit des cures non aplasiantes réalisables en ambulatoire (cures d’entretien). Chez les sujets de moins de 60 ans, une chimiothérapie de consolidation comportant deux à trois cures aplasiantes étalées sur une période de quatre à six mois est optimale pour prévenir la rechute. Les chimiothérapies hyperintensives accompagnées ou non d’irradiation corporelle totale et suivies de greffe de cellules souches hématopoïétiques autologues semblent donner des résultats égaux ou légèrement supérieurs à ceux de la chimiothérapie de consolidation. En cas d’impossibilité de réaliser une chimiothérapie intensive du fait de sa toxicité, un traitement d’entretien sur une période de 12 à 18 mois est généralement réalisé.
2 - Allogreffe de CSH :
Elle représente le traitement de post-induction le plus efficace pour prévenir la rechute. Elle n’est réalisable que chez les sujets jeunes (moins de 50 ans) à partir d’un frère ou d’une soeur HLA identique. Ces deux conditions font que ce traitement est actuellement pratiqué chez moins de 10% des patients atteints de leucémie aiguë myéloblastique. Actuellement, de nouvelles méthodes d’allogreffe utilisant un traitement immunosuppresseur, une greffe de cellules souches hématopoïétiques sanguines et une immunothérapie cellulaire (injection de lymphocytes de donneur) secondaires sont en cours d’évaluation.
C – PRONOSTIC :
Les facteurs suivants correspondent généralement à un pronostic plus défavorable: antécédents de maladie hématologique, leucémies secondaires, âge supérieur à 65 ans, état général précaire lors du diagnostic, hyperleucocytose initiale, M5, difficultés d’obtention de la rémission.
La chimiothérapie permet d’obtenir une rémission complète dans 70 à 80% des cas chez les sujets jeunes. Ce pourcentage diminue à 50% au delà de 60 ans. La survie à 5 ans des patients chez qui une rémission complète est obtenue peut être évaluée entre 25 et 50% après consolidation chimiothérapique ou autogreffe. Le taux de rechute atteint 50 à 70%. La survie à 5 ans après greffe de moelle allogénique est de 50 à 70% avec un taux de rechute de 15 à 20%. Chez les sujets de plus de 60 ans chez qui seule une chimiothérapie d’entretien peut être entreprise la survie à 5 ans n’excède pas 15 à 20%.
Conclusion
LAM : pathologie grave
Diagnostic souvent facile (frottis sanguin + myélogramme)
Situations difficiles:
immunomarquage ,cytogénétique aide +++
Marqueurs Pc
Collaboration étroit entre clinicien et biologiste
LEUCEMIE AIGUË MYELOBLASTIQUE(LAM)
 Reviewed by Admin
on
23:32:00
Rating:
Reviewed by Admin
on
23:32:00
Rating:
Reviewed by Admin
on
23:32:00
Rating:






{kind=link}
{kind=link}
Aucun commentaire: