


INTRODUCTION
oThrombocytémie essentielle (TE) est un Syndrome
myéloprolifératif caractérisé par une thrombocytose stable
(PLT≥450 G/l) et une prolifération excessive de mégacaryocytes.
oClinique: manifestations thrombotiques et hémorragiques.
oDiagnostic de la thrombocytémie essentielle est un
diagnostic d’élimination.
EPIDEMIOLOGIE
Incidence: 0.6-2.5 cas/100 000/an (Polycythemia Vera StadyGroup)
TE atteint de façon égale H et F 50-60 ans, peut survenir à
n’importe quel âge rarement chez l’enfant. (à différentier avec les
cas rares de thrombocytose héreditaire)
TE atteint de façon égale H et F 50-60 ans, peut survenir à
n’importe quel âge rarement chez l’enfant. (à différentier avec les
cas rares de thrombocytose héreditaire)
PHYSIOPATHOLOGIE
La TE est caractérisée par une prolifération du tissu
hématopoïétique médullaire intéressant surtout la lignée
mégacaryocytaire.
Maladie monoclonale de la CSH (75%) étudiée par
méthodes de biologie moléculaire et cytogénétique.
Formation de colonies MGC (70%) et/ou érythroïdes (60%)
spontanées (in vitro) par les progéniteurs mégacaryocytaires
et érythroblastiques.
Un taux normal de l’ IL-6 normal (contre les
hyperplaquettose où le taux de l’IL6 est élevé.)
40 à 50 % JAK2V617F => prolifération indépendante
des facteurs de croissance (notion de charge de l’allèle muté).
Mutation de MPL, MPL W515K/L reportée dans 1%.
20% surexpression du gène PRV-1 au niveau des granulocytes
Anomalies de l’hémostase:
Nombre de grains denses diminué (sérotonine et ADP).
Thrombopathie: hypoagrégabilité à l’adrénaline, à l’ADP, au
collagène ou une aggrégation après un temps de latence prolongé.
Diminution des inhibiteurs naturels de la coagulation (AT,PS, PC).
ASPECTS CLINIQUES
Découverte de la maladie peut être fortuite à l’occasion d’un
hémogramme systématique et les 2/3 des patients sont
asymptomatiques
Occlusions microcirculation sont les plus fréquents:
Crisesd’érythromélalgis
Ulcères de jambe
SNC: cécité, paresthésies, hémiparésie.
Erythromélalgie:
le signe le plus fréquent(60% des cas) caractérisé par des douleurs
du type brûlure avec rougeur des orteils et de la plante des pieds ou
doigts et de la paume des mains, calmées par l’aspirine.
Thromboses artérielles (15-20%)++:
ischémies myocardiques, cérébrales, rétiniennes. Parfois veineuses:
mbres inf, Vaisseaux portes, spléniques ou mésentériques.
Signes hémorragiques ce sont les plus rares, provoqués ou
spontanées:
ecchymoses, épistaxis, hémorragie après extraction dentaire ou
intervention chirurgicale.
SMG 30% généralement modérée.
DIAGNOSTIC BIOLOGIQUE
Hémogramme:
Pas d’anémie (sauf hémorragie)
GB modérément élevé: 10-20 G/l,
polynucléose sans myélémie
PLT > 450 G/L voire > 1000 G/l.
Frottis sg: amas PLT, anisocytose PLT
(PDW élevé), PLT géantes,
Etude MO
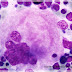
Myélogramme:
MO très riche avec:
Mégacaryocytes nombreux, polymorphes, de grande
taille et à noyau hyperlobulé d’aspect en bois de cerf:
staghorn (éliminer une myélodysplasie).
BOM:
Hyperplasie médullaire globale avec hyperplasie et
dystrophie de la lignée mégacaryocytaire, souvent en
amas, de grande taille à noyau hyperlobé.
Pas de réticulofibrose, ni de prolifération associée des
lignées granuleuse ou érythroblastique
hématopoïétique médullaire intéressant surtout la lignée
mégacaryocytaire.
Maladie monoclonale de la CSH (75%) étudiée par
méthodes de biologie moléculaire et cytogénétique.
Formation de colonies MGC (70%) et/ou érythroïdes (60%)
spontanées (in vitro) par les progéniteurs mégacaryocytaires
et érythroblastiques.
Un taux normal de l’ IL-6 normal (contre les
hyperplaquettose où le taux de l’IL6 est élevé.)
40 à 50 % JAK2V617F => prolifération indépendante
des facteurs de croissance (notion de charge de l’allèle muté).
Mutation de MPL, MPL W515K/L reportée dans 1%.
20% surexpression du gène PRV-1 au niveau des granulocytes
Anomalies de l’hémostase:
Nombre de grains denses diminué (sérotonine et ADP).
Thrombopathie: hypoagrégabilité à l’adrénaline, à l’ADP, au
collagène ou une aggrégation après un temps de latence prolongé.
Diminution des inhibiteurs naturels de la coagulation (AT,PS, PC).
ASPECTS CLINIQUES
Découverte de la maladie peut être fortuite à l’occasion d’un
hémogramme systématique et les 2/3 des patients sont
asymptomatiques
Occlusions microcirculation sont les plus fréquents:
Crisesd’érythromélalgis
Ulcères de jambe
SNC: cécité, paresthésies, hémiparésie.
Erythromélalgie:
le signe le plus fréquent(60% des cas) caractérisé par des douleurs
du type brûlure avec rougeur des orteils et de la plante des pieds ou
doigts et de la paume des mains, calmées par l’aspirine.
Thromboses artérielles (15-20%)++:
ischémies myocardiques, cérébrales, rétiniennes. Parfois veineuses:
mbres inf, Vaisseaux portes, spléniques ou mésentériques.
Signes hémorragiques ce sont les plus rares, provoqués ou
spontanées:
ecchymoses, épistaxis, hémorragie après extraction dentaire ou
intervention chirurgicale.
SMG 30% généralement modérée.
DIAGNOSTIC BIOLOGIQUE
Hémogramme:
Pas d’anémie (sauf hémorragie)
GB modérément élevé: 10-20 G/l,
polynucléose sans myélémie
PLT > 450 G/L voire > 1000 G/l.
Frottis sg: amas PLT, anisocytose PLT
(PDW élevé), PLT géantes,
Etude MO
Myélogramme:
MO très riche avec:
Mégacaryocytes nombreux, polymorphes, de grande
taille et à noyau hyperlobulé d’aspect en bois de cerf:
staghorn (éliminer une myélodysplasie).
BOM:
Hyperplasie médullaire globale avec hyperplasie et
dystrophie de la lignée mégacaryocytaire, souvent en
amas, de grande taille à noyau hyperlobé.
Pas de réticulofibrose, ni de prolifération associée des
lignées granuleuse ou érythroblastique

Aspect histopathologique

Classification OMS 2008
1. Augmentation persistante des plaquettes 450
x109/L
2. Biopsie médullaire:
prolifération mégacaryocytaire prédominante
a/ majorité d’éléments mûrs et de grande taille.
b/ pas d’augmentation significative de
la granulopoïèse neutrophile ni de l’érythropoïèse
c/ pas d’excès d’éléments immatures dans ces deux
lignées.
3. Absence des critères de
a/PV b/ PMF c/ LMC d/ MDS e/autre
maladie maligne de la lignée myéloïde.
4. mutation JAK2V617F ou autre marqueur de
clonalité, en l’absence de marqueur de clonalité :
absence d’argument en faveur d’uneThrombocytose
Réactionnelle.
DIAGNOSTIC DIFFERENTIEL
oThrombocytose réactionnelle: Carence martiale
Perte hémorragie (aiguë ou chrq) Splénectomie ou
hyposplénisme
Néoplasies
Inflammation chrq
(vacularite, connectivite,
PR, infections chrq)
Thrombocytose de réparation (TRT PTI, AP
chimiothérapie)
oThrombocytose accompagnant un autre SMP
,LMC,PV,MF
oThrombocytose associée à un SMD
oThrombocytose familiale: mutation gène TPO
x109/L
2. Biopsie médullaire:
prolifération mégacaryocytaire prédominante
a/ majorité d’éléments mûrs et de grande taille.
b/ pas d’augmentation significative de
la granulopoïèse neutrophile ni de l’érythropoïèse
c/ pas d’excès d’éléments immatures dans ces deux
lignées.
3. Absence des critères de
a/PV b/ PMF c/ LMC d/ MDS e/autre
maladie maligne de la lignée myéloïde.
4. mutation JAK2V617F ou autre marqueur de
clonalité, en l’absence de marqueur de clonalité :
absence d’argument en faveur d’uneThrombocytose
Réactionnelle.
DIAGNOSTIC DIFFERENTIEL
oThrombocytose réactionnelle: Carence martiale
Perte hémorragie (aiguë ou chrq) Splénectomie ou
hyposplénisme
Néoplasies
Inflammation chrq
(vacularite, connectivite,
PR, infections chrq)
Thrombocytose de réparation (TRT PTI, AP
chimiothérapie)
oThrombocytose accompagnant un autre SMP
,LMC,PV,MF
oThrombocytose associée à un SMD
oThrombocytose familiale: mutation gène TPO
EVOLUTION-PRONOSTIC
oSurvie de 95% à 10 ans
oEvolution chronique pendant 10-20 ans, parfois
des complications:
Manifestations hémorragiques:
hgies mineures, majeures si PLT > 1500 G/L
(déficit acquis en vWF avecperte des multimères de
haut poids moléculaire).
Les complications thrombotiques: artères périph
(37%), cerveau (31%), cœur (23%).
Risque patients > 60ans ou avec autre facteur de
risque (tabagisme).
Complications hématologiques:
Myélofibrose assez fréquente, LA de type granulo
monocytaire (Rare). Complications iatrogènes :
type d’aplasie médicamenteuse ou le risque de
favoriser la survenue d’une LA induite.
oEvolution chronique pendant 10-20 ans, parfois
des complications:
Manifestations hémorragiques:
hgies mineures, majeures si PLT > 1500 G/L
(déficit acquis en vWF avecperte des multimères de
haut poids moléculaire).
Les complications thrombotiques: artères périph
(37%), cerveau (31%), cœur (23%).
Risque patients > 60ans ou avec autre facteur de
risque (tabagisme).
Complications hématologiques:
Myélofibrose assez fréquente, LA de type granulo
monocytaire (Rare). Complications iatrogènes :
type d’aplasie médicamenteuse ou le risque de
favoriser la survenue d’une LA induite.
MODALITES THERAPEUTIQUES
oPrévention des complications: Diminution du
chiffre des plaquettes réduise la survenue de
complication thrombotique.
oTRT sujets à haut risque:
Âge > 60 ans
ATCD de thrombose
PLT > 1000 G/L
Facteurs de risque: diabète, HTA
=>Hydroxyurée+aspirine/anagrélide( agent
thrombopéniant par inhibition de la
prolifération et la différenciation des
mégacaryocytes)+aspirine /IFNα.
oPatients à risque intermédiaire:
40-50 ans
Pas autre facteur de risque
Aspirine /Hydroxyurée+aspirine
oPatients à risque faible:
<40ans
Pas d’autre fact de risque
Aspirine faible dose (si CI:
dipyridamole, ticlopidine…)
oFemme enceinte: aspirine à 100mg/j, IFNα
chiffre des plaquettes réduise la survenue de
complication thrombotique.
oTRT sujets à haut risque:
Âge > 60 ans
ATCD de thrombose
PLT > 1000 G/L
Facteurs de risque: diabète, HTA
=>Hydroxyurée+aspirine/anagrélide( agent
thrombopéniant par inhibition de la
prolifération et la différenciation des
mégacaryocytes)+aspirine /IFNα.
oPatients à risque intermédiaire:
40-50 ans
Pas autre facteur de risque
Aspirine /Hydroxyurée+aspirine
oPatients à risque faible:
<40ans
Pas d’autre fact de risque
Aspirine faible dose (si CI:
dipyridamole, ticlopidine…)
oFemme enceinte: aspirine à 100mg/j, IFNα
CONCLUSION
oTE=> Dc d’exclusion
oCas JAK2+:
Perspective: TRT ciblé
oCas JAK-: autres mutations à découvrir
oCas JAK2+:
Perspective: TRT ciblé
oCas JAK-: autres mutations à découvrir
Thrombocytémie Essentielle
 Reviewed by Admin
on
23:45:00
Rating:
Reviewed by Admin
on
23:45:00
Rating:
Reviewed by Admin
on
23:45:00
Rating:




")


Aucun commentaire: